See Your ROI with Greenlight Guru in Seconds


Experience the #1 QMS software for medical device companies first-hand. Click through an interactive demo.


In today’s highly connected world, medical devices often include state-of-the-art software, or, the standalone software itself is considered a medical device.
Software as a Medical Device (SaMD) is a niche area experiencing a rapid increase in use and, therefore, is receiving some attention from regulatory bodies. Regulations and guidelines are being updated to keep up with the new technology.For medical device companies, there is a common confusion over requirements specifically related to SaMD regulation. I recently sat down with Andrew Wu, a Software Consultant for Rook Quality Systems to discuss this topic on air while recording our weekly podcast. In his role, he ensures that SaMD companies have the proper systems and processes in place to meet regulatory requirements.
Here’s what Andrew recommends you know when it comes to SaMD:
Free Resource: Download this PDF and learn what influences SaMD classification.
SaMD is a standalone software that performs one or more medical purposes without being a part of a hardware device. This is a term that has been defined directly from the FDA, as its use continues to increase.
Let’s take a look at a few examples to determine whether or not these devices would classify as a SaMD:
The FDA published a guidance back in 1997 governing software in medical devices, but the landscape has significantly changed over the last couple decades. This foundational piece of regulation is becoming obsolete due to the emergence of innovative software. As a result, the FDA is taking steps to address this shortfall.
For example, the FDA is now offering a pre-certification program for digital health applications. This allows developers or manufacturers to create, adapt or expand the functionalities of their software. They are also able to maintain a dialogue with the FDA, ensuring they’re on the right track, while still being able to make timely changes as needed. This is a good example of how the FDA is working in a progressive way to better align regulatory programs with development needs.
It’s very common amongst software teams to not be fluent in “regulatory” language. They’re brilliant developers, but they don’t know the medical device terminology and the regulations can oftentimes be written in cryptic and seemingly contradictory terms.
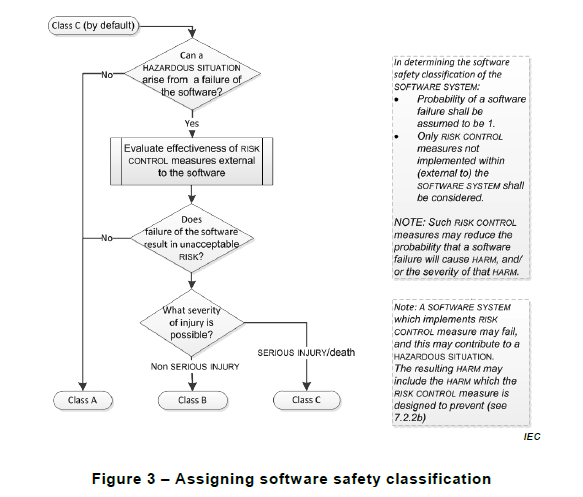
Somewhere along the way, they stumble upon the IEC 62304 standard, but there’s still a disconnect with actually being able to apply it. One of the big confusions happens around classification. This should come at no surprise since step one of the regulatory process involves identifying the classification of your product.
The 62304 standard is an ISO consensus standard providing guidance for software manufacturers on how they should manage their life cycles, from development to deployment and post-market surveillance. This standard has been widely adopted and agreed upon by many regulatory bodies across the world. And of note, adherence to standards is not mandatory.
Early guidance around classification (from the early 2000’s) was somewhat unclear. It was left up to manufacturers to determine and prove their own class. However, around 2005 there was an amendment to 62304 resulting in the publication of IEC 62304:2016, which provides a helpful flow chart for manufacturers to follow when identifying the class of their medical device.
This helps manufacturers to delineate by hazards and the risk measures they plan on developing. They also consider the severity of possible injuries in identifying software class. But realize the software class does not necessarily relate to regulatory product classification system.
There are two other resources besides 62304 that are valuable to look at when considering software classification. These are the FDA's Level of Concern, which rates concern from minor to major, and the SaMD classification and guidance from the International Medical Device Regulators Forum.
It’s important to understand how each of the regulatory bodies have determined risk structure and you should connect with others who have gone through this process before. You need to understand which information to provide and the impact of its safety profile when a failure or defect occurs.
Here’s a quick rundown of the varying classifications by regulatory body:
IEC 62304 standard:
FDA's Level of Concern:
You should do your due diligence to find out whether there are any predicate devices that have already gone to market, akin to your own, so you can take advantage of the regulatory pathways other manufacturers have taken. The bottom line is, you need to build your best strategy to mitigate the chances of ending up in a risky situation.
Device classification involves a critical understanding the scope of documents you’re expected to provide. For example, if you look at the documentation requirements for the FDA’s Level of Concern, Minor concern devices require very little documentation (compared to Major concern) for the FDA to review. With that said, you still need to have them readily available in your quality system and maintain traceability.
Let’s look at the same example, but on the other end of the Level of Concern spectrum. Suppose your SaMD has a Major concern, the FDA will require you to provide an exorbitant amount (more than what’s actually needed, but that’s the point) of documentation to help them review the device. This may include requests for post-market surveillance data on performance, or any other information they find relevant and determine should be addressed.
Device manufacturers should be in constant contact with the FDA and experienced consultants about regulations because they’re frequently being updated and/or modified. Establishing communications lines early on with these professionals will be beneficial, not just for building a strategic relationship, but for building protective checkpoints into the foundation of your product.
Don't miss these free tips: Learn about What Influences SaMD Classification here.
SaMD companies need to have a good quality management system (QMS) during the development process to ensure compliance with any and all regulations; don’t wait until you’re in damage-control mode and it’s too late! It’s always much more difficult to scramble together a QMS after the fact. Whichever QMS platform you choose, make sure it’s capable for compliance with applicable regulatory requirements, such as FDA 21 CFR Part 820 and ISO 13485:2016.
Adopting a formal QMS early in the development process will be instrumental in managing the meticulous documents and records you’ll need easy access to, both for internal purposes and as required by regulatory bodies. Companies often tend to learn best by doing, including making key mistakes with quality management. Following a good QMS framework with help from experienced people can help not only with the quality of the product but the likelihood of regulatory success.
Remember, if you are a developer of a SaMD, you are considered a manufacturer by definition, even though you’re making a software product! Regulatory language that includes any use of the word “manufacturer” applies to you!
Another common objection I’ve heard is somewhere along the lines of; “Oh, well that FDA stuff is for waterfall development. We’re agile so we can’t really do that.” To set the record straight, nowhere in any regulatory standard does it prescribe that a waterfall methodology needs to be used.
The methodology or approach you choose is entirely up to you to decide, but you simply must define it clearly in a procedure. You may take any approach you believe best compliments the development efforts of your unique operations, as long as you document your activities and demonstrate traceability of design control and risk management elements.
In closing, I have only a couple points I’d like to reiterate and that’s to be familiar with your software classification and adhere to the associated requirements. For an out-of-the-box QMS that’s automatically programmed to keep you compliant with medical device regulations and in sync with your internal communications, Greenlight Guru, has you covered.
Streamline your compliance by getting your free demo ➔