See Your ROI with Greenlight Guru in Seconds


Experience the #1 QMS software for medical device companies first-hand. Click through an interactive demo.


Design controls and risk management processes should be tools for MedTech professionals to ensure that medical devices are designed, developed, and manufactured to be safe, effective, and address indications for use.
All too often, however, design controls and risk management are viewed as a pile of “stuff” FDA and other regulatory bodies require during the design and development process.
FDA design control regulations have now been in place for 20 years. They should be aids to guide MedTech professionals, but an overwhelming percentage of the medical device community approaches these activities as checkbox, low-value-add tasks.
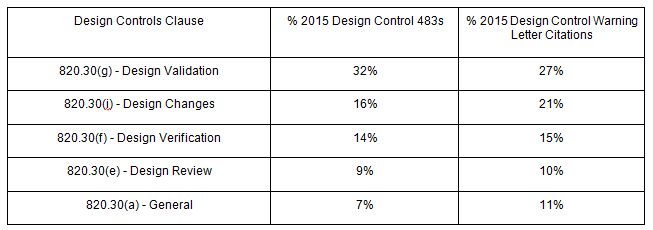
Year after year, design control deficiencies rank as the single biggest reason medical device companies receive 483 observations; such deficiencies also rank in the top three reasons for warning letters.
Despite “risk management” only being officially cited as part of 820.30(g) - Design Validation, FDA requires medical device companies to implement risk management processes to analyze, evaluate, assess, and control risks throughout the entire product lifecycle.
In fact, according to 2015 FDA data, 32 percent of design control 483 observations and 27 percent of design control warning letter citations relate to 820.30(g).
Most, if not all, of the above FDA actions were avoidable. Let’s dive deeper into why your design controls and risk management processes may be failing, and discuss what you can do about it.
Bonus Giveaway: Click here to download a free Risk Management Plan Template.
A medical device product development process needs to establish the major stages, deliverables, and milestones from project initiation through market launch. Design controls and product risk management should be integrated within that development process.
However, medtech companies commonly err in one direction or another during product development, making the process too detailed and overly burdensome, or too vague (or maybe, not defined at all).
The overly burdensome process is difficult to follow. Too many tasks and actions are required. Following a lengthy, detailed process often presents numerous opportunities for non-conformances and increases risks for non-compliances. The vaguely defined process is also problematic. Lack of clearly defined stages and deliverables leads to possible non-conformances and non-compliances with regulatory expectations.
Your product development process needs to be “right-sized,” according to the type and size of your company, as well as the types of medical devices you design and develop. Use 21 CFR 820.30 and ISO 13485 (section 7.3) as a guide to determine minimum design control deliverables requirements.
Use the requirements of ISO 14971 to identify minimum risk management requirements. Then, build your product development process around the design controls and risk management requirements you’ve defined.
If your company has additional business needs, add these to your process, erring on the side of minimal deliverables.
Medical device development often is triggered by a desire to address unmet clinical needs, or to improve upon current treatment options. Identifying a clinical issue to solve lays the groundwork for defining intended use and indications for use for your medical device.
Often, a prototype is involved in the process at this stage, used to help further define and articulate user needs. For many product development engineers, this early discovery is one of the most thrilling parts of the medical device process — so much so that it is common to go full speed ahead once the prototype seems to be sufficient and satisfactory, even though the end user’s needs may not be fully understood.
Who is the “end-user”? Is it the patient? Is it the clinician? Usually, “yes” answers all of those questions; these are the easy users to identify. But, are there other users? Typically, yes. End-users can include distributors, doctors, nurses, biomed technicians, patients, patient caregivers, reprocessing personnel, and so on
Consider all the steps that occur between a medical device leaving your facility, through point of use, and ending when the product is no longer is use. Each end user likely has a list of needs, each of which you need to document. User needs feed the design and development process and are the precursor to establishing your device’s design input requirements.
Design inputs are the most important part of product development, establishing the foundation of a medical device.
Often, there is a rush to get through the design inputs stage so that additional prototypes can be built, tested, etc. I get it. Just about every product development project I have ever worked on had the target completion date defined before the project ever officially began, so getting everything done sometimes seemed to take precedence.
But design inputs are a continuation of user needs, as well as a precursor to design verification. When defining design inputs, you need to consider exactly how the design outputs will confirm design inputs have been met. By rushing through design inputs, you risk producing requirements that are not readily verifiable.
To avoid this scenario, use your desire to prototype to your advantage. Mini tests or experiments performed with your prototypes can help determine how to verify.
Many medical device product developers engage in formal product risk management activities way too late in the process. The right time to start documenting risk management activities is when you are defining user needs and design inputs, so that identified risks can guide those requirements, as well as improve your ability to manage design verification and validation.
Understanding possible hazards, hazardous situations, and possible harms will help you improve your medical device design. By incorporating risk management earlier, you will be able to leverage later stage product development activities, such as design verification and validation, to support risk-based decision-making regarding product development.
Design reviews are important opportunities, conducted at appropriate stages throughout product development, to assess progress with respect to overall product design and development.
Design reviews should be constructive and introspective, and design control activities should be included and reviewed. Design review attendees should vary, depending on the design review’s focus during a particular development stage. Common design review mistakes include a lack of review documentation, failure to represent all appropriate functional areas, and/or omission of objective evidence verifying that action items identified during design reviews have been completed.
Another common issue is trying to cram too much into a single design review. While, technically, this might be considered acceptable (provided it aligns with your design and development plan), a design review that includes too many design controls and product development stages is likely to add little overall value and be ineffective. Plus, more information included within a design review will correlate to a longer meeting and more action items.
I recommend having more frequent design reviews, each focusing on a finite set of information, and trying to limit your design review to under an hour.
Design verification will only be as good as the design input requirements. While that connection is clear, keep in mind that design outputs also contribute to successful design verification.
Design verification demonstrates that the design outputs meet the design inputs, serving as proof that you designed your product correctly. Your design verification acceptance criteria may be captured as part of design outputs or inputs, and you must ensure that acceptance criteria has been defined before conducting verification.
Finally, testing is considered to be synonymous with design verification. While testing is an acceptable method for conducting design verification, so are inspection and analysis.
Design validation should demonstrate that the medical device meets the needs of the end user, who must be clearly defined and identified. Failure to do so can lead to design validation that is haphazard and misses the mark.
Furthermore, design validation needs to involve those end-users, and products built for design validation need to be from production-equivalent materials and processes.
Chances are, your medical device product will change. The design may change, or the manufacturing processes may change. If so, you need to document these changes and ensure that the design controls are updated accordingly. Additionally, you likely will need to have design reviews for the changes.
Even after the medical device it is transferred to production, changes may continue to happen, making design controls a continuous process.
Sometimes, this is not clear: Conventional wisdom states that design controls are a set of historical records describing design and development, and then archived after design transfer. But, any changes you make to a medical device in production / post-production require design controls.
You also need to determine if verification and/or validation is required for your design change, as well as evaluate the impact on user needs and design inputs. The design outputs you established during product development now become the basis of the product device master record.
Thus, design controls are a continuous, total product lifecycle process. As the medical device world continues to move toward risk-based approaches regarding quality management systems, I would expect there to be more scrutiny when it comes to product lifecycle risk management, too. By keeping these two ideals at the forefront of your product development process, you both insulate yourself from regulatory repercussions and better your chances of market success.
This article originally appeared as a guest expert post on Med Device Online.









